
Learning Objectives This week, students will:
- work with newick Files
- access and list the elements of a
phyloobject- obtain phylogenetic data from the Open Tree of Life
Practice Objectives
- Exploring the structure of an object
- plot phylogenies with
ape::plot.phylo()andggtree::ggtree()- customize phylogenetic plots with
ggtree::ggtree()- understand the relationship between lists and the
phyloclass as data structures.Non Objectives
- simulate phylogenies
- get dated phylogenies using
datelife
Setup your RStudio project (5 min)
- 🎗️ Structuring your files into a project is a best practice for good data science!
- Open your RStudio project for the class; I called mine “spring2023”.
- Open a new Rmd file, name it “trees-opentree.Rmd”, and save it to your “documents” folder.
- load the packages
rotlandape- the name of the package
apeis an acronym that stands for Analysis of Phylogenetics and Evolution - it was mainly developed for the analysis of molecular sequences
install.packages("ape")

- the name of the package
- the name of the package
rotlstand for an R inrerface to interact with the OpenTree of Life.
Review (10 min)
- What is a phylogeny?
- A phylogeny is a hypothesis of ancestor-descendant relationships (aka evolutionary relationships)/
- We represent this hypothesis graphically in the form of a tree:

- The main parts of a phylogenetic tree are:
- Tips, represent our observations; either a living species, a fossil, a sample of a virus, etc.
- Nodes, represent a hypothesis of common ancestry. That means, based on evidence, we think that two or more lineages share a common acestor some time in the past; or that two or more lineages descend from the same lineage and diverged from it some time in the past.
- Branches, represents a measure of amount of change that occured between lineages in a measure of time (commonly known as evolutionary distance).
- branches by themselves cannot say in wich direction the change occurred.
- Root, is a special type of node, as it represents the common ancestor to all lineages in the tree.
- The position of the root provides a direction of evolution.

Both of these phylogenetic trees shows the relationship of the three domains of life (Bacteria, Archaea, and Eukarya), but the (a) rooted tree attempts to identify when various species diverged from a common ancestor, while the (b) unrooted tree does not.
Caption from original Figure 20.1𝐴.1 of LibreTexts
- Discussion: How do we construct phylogenetic trees?
- How long does it take?
- Etherpad quiz: How hard it is to get a phylogenetic tree? How much time do you need to get a “good” phylogenetic tree?
The Open Tree of Life platform (10 min)
- Overview of the OpenTree website (5 min)
- OpenTree’s Web interface
- Phylogenetic Database (Phylesystem)
- Curation
- Downloading trees in newick format
Reading trees into R with read.tree()(10 min)
read.tree()function from the packageape- Usage to read a phylogeny from a newick file in your computer:
- provide the path and file name
- 🎗️ Use relative paths!
ape::read.tree("relative/path/newick-file-name.tre")
- Usage to read a phylogeny from a URL address:
- Provide the URL address as a character vector:
ape::read.tree("url_adress_ of_newick_file.tre")
- Provide the URL address as a character vector:
- Example:
- Read into R the newick tree in the file “portal-tree.tre”
- create an object named
opentree_tree:opentree_tree <- read.tree(file = "../data-raw/tree-from-opentree.tre") - Read into R the newick tree from URL “http://ape-package.ird.fr/APER/APER2/primfive.tre”,
- To copy the URL, go to
ape’s book website and copy the link to primfive.tre - create an object named
small_tree:small_tree <- read.tree(file = "http://ape-package.ird.fr/APER/APER2/primfive.tre")
Plot a phylogeny with the package ape (5 min)
plot.phylo()function from the packageape:plot.phylo(opentree_tree) plot.phylo(small_tree)- plotting phylogenies with
aperequires a lot of customization. - we will not cover that in this course,
Getting trees with specific taxa from OpenTree
- Difference between phylogeny and taxonomy (5 min)
- What is taxonomy?
- The science of description, identification, nomenclature, and classification of organisms.
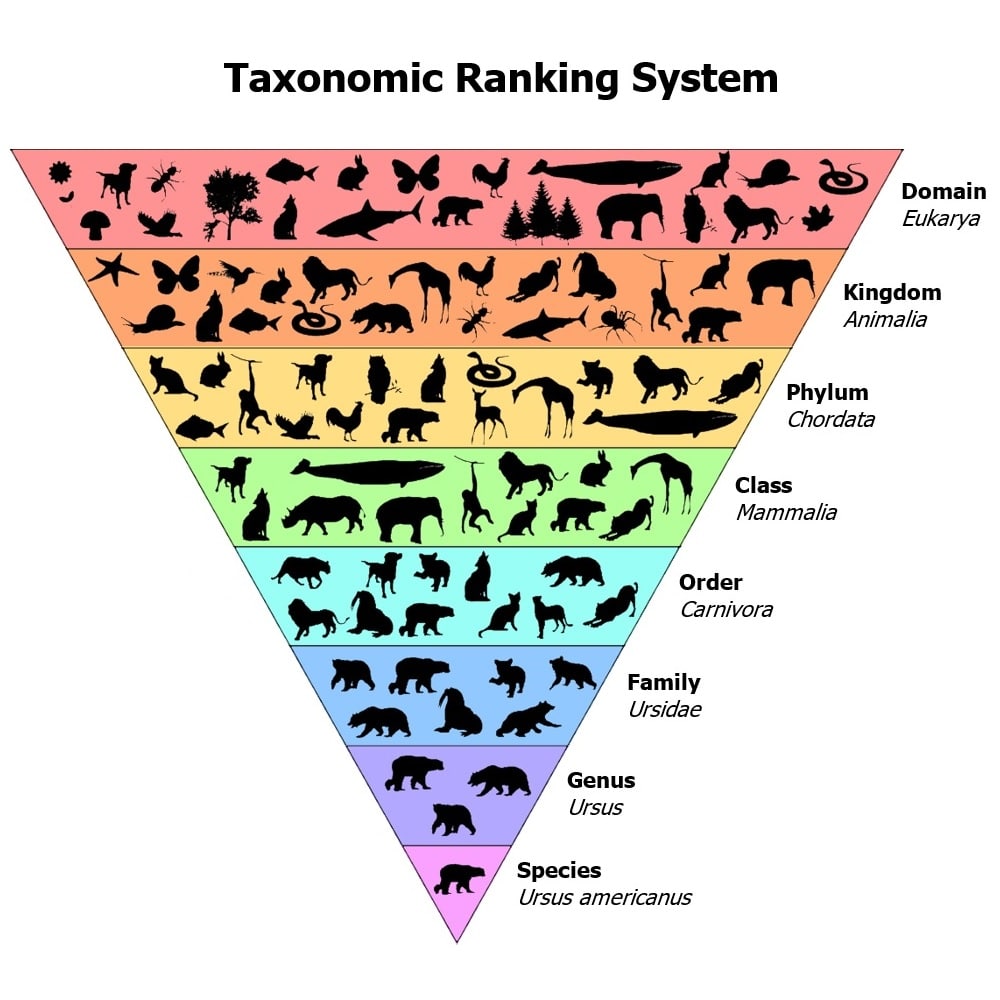
In a broad sense, taxonomy is the method used for organizing similar content into relevant groups. To put it even more broadly, taxonomy is how we classify things. From its conception, taxonomies have played an important role in biological science, where it has been largely used to organize the animal kingdom. Think of mammals vs. birds vs. reptiles and all the details in between: within the mammals group, we have cats, whales, apes, etc.; as we move further down the line, we have different species of apes such as gorilla, chimpanzees, etc. If you can visualize this as a tree of sorts, you’re already on the way to understanding what a taxonomy is at its basic level.
Text from Taxonomy management 101.
- Finding your taxa in the Open Tree of Life Taxonomy
-
Go to https://tree.opentreeoflife.org/opentree/argus/opentree13.4@ott93302 and look for a group that your are interested in or that is in your data set that you are going to analyze for the final project
- Getting OTT ids for a taxon with the function
tnrs_match_names() - Exploring the
match_namesobject - Extracting data from a
match_namesobject - Getting a piece of the Synthetic Open Tree of Life
- The synthetic Open Tree of Life (synthetic OpenTree from now on) summarizes information from 1239 trees from 1184 peer-reviewed and published studies, that have been uploaded to the OpenTree database (the Phylesystem) through a curator system.
tol_about()
- Getting a subtree of one taxon with
tol_subtree() - Example: Get a subtree of all amphibians